

How to survive an FDA inspection! Part 1
It’s a Friday, late morning or early afternoon. You’re in the conference room with a few other executives of your medical device company. Also in the room is an inspector from the US Food and Drug Administration, the FDA. The inspector showed up the previous Monday for a scheduled inspection, which has taken all week. The inspector is now going to review the findings of the inspection.
How do you feel about this? Are you worried or concerned? If so, why? Or do you have a high degree of confidence in your QMS and your people, and so you feel it will go well? There’s no reason you can’t be confident if you’ve properly prepared. The good news is, the FDA publishes documents that will allow you to do exactly that.
You have a QMS. Either you are using a commercially available software package, a QMS prepared for your company by a qualified regulatory consulting firm, or your internal team has developed its own compliant system. In any of these cases, the QMS has presumably been prepared with exacting attention to the detail needed for your specific company’s operations.
So what can your company do to gain confidence? Perform a mock FDA inspection periodically and do it the way the inspector is going to!
How do I know what the inspector is going to ask or look for? Follow the same guidance documents that the inspectors use. The key ones include:
- Guide for Quality Systems Inspections (QSIT)
- FDA Compliance Guidance Manual – Inspection of Medical Device Manufacturers
- IOM – Complete Investigations Operations Manual
Study these documents, starting with the QSIT and establish a program to conduct periodic internal quality audits following these provisions. In other words, duplicate an FDA inspection periodically. Be appropriately critical of your own company and “write up” internal 483 like observations along with proposed corrective actions. Then follow up with those corrective actions and document their adequate completion.
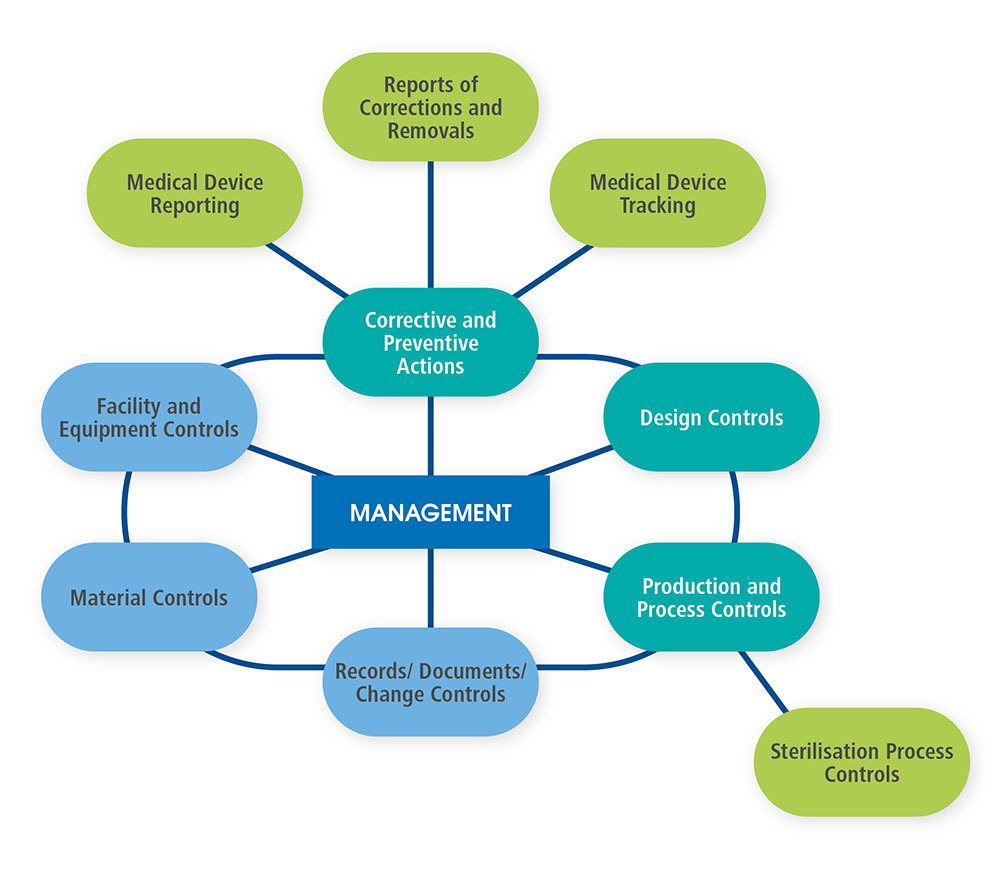
Just behind the cover page of the QSIT, the FDA provides a diagram of the interactions between management and several operational aspects of a company following a Quality Management System. That diagram is shown below as Figure 1 and redrawn for improved readability.
Figure 1.
Below are several key items, common questions, and excerpts from the documents.
Q. Will the FDA just show up, or will they contact me in advance?
A. In almost all circumstances, especially for routine inspections, you will be contacted in advance and told when the FDA will come for the inspection.
Here’s the excerpt on this topic from the IOM:
Pre-announcements are mandatory for all medical device surveillance inspections.
The pre-announcement should generally be no less than 5 calendar days in advance of the inspection. Should a postponement be necessary, the decision as to rescheduling rests with the investigator/team, but the new inspection date should not be later than 5 calendar days from the original date.
Q. Are there different kinds of inspections?
A. Yes. Here are some excerpts from the IOM.
- Pre-market inspections (PMA, 510(k))
- Foreign inspections
- Instructed by Compliance Program, assignment or directive
- Quality System Surveillance Inspections
Types of Inspections:
- Initial – first inspection
- Routine – normal surveillance inspection
- Follow-up – follow-up to a violative inspection
- For-cause – inspection to follow-up on a specific issue
- Other (please specify) – inspection that doesn’t meet one of the other categories (will be used very rarely)
Q. How long will the inspection take?
A. For routine Quality System Surveillance Inspections, the FDA usually plans for one week. Starting on a Monday and finishing on Friday is a typical example. The process may take longer if they’re looking for specific details regarding a Medical Device Report (e.g., the inspection is “For-cause,” or if they’re exploring issues where potential violations or non-compliance is suspected.
Q. How do I know the people requesting to inspect my operation are legitimate?
A. Here’s the excerpt from the IOL:
Introduce yourself by name, title and organization. Show your credentials to this person and present a properly signed, completed original of the FDA 482, Notice of Inspection
Like most business organizations, the FDA has a lot to do and organizes in a way to complete their tasks in a structured, efficient manner. There are two primary approaches to a Quality System Surveillance Inspection: Top Down and Bottom Up.
Top-Down inspections begin with your QMS, your quality manual, quality policy, etc. in this case the inspector will most likely follow the QSIT guidelines looking into the four major areas of interest along with select sub-topics as described below. The inspector will follow procedures, documentation, and records down into specific products or issues.
Bottom-Up inspections start with a specific issue or product. The records and procedures related to that specific issue or product are examined in detail and the various pathways followed into higher, oversight documents.
Here’s a statement about these approaches from the QSIT:
“One similarity between “top-down” and “bottom-up” inspectional approaches is record review. Both approaches involve review of raw data, or individual records.
The organization of initial, routine Quality System Surveillance Inspections follows a structure than looks at key portions of the quality system. Here’s a description of that approach excerpted from the QSIT:
Rather than check every aspect of the firm’s quality system, the subsystem approach focuses you on those elements that are most important in meeting the requirements of the quality system regulation and which are key quality indicators.
We have chosen four major subsystems that are the basic foundation of a firm’s quality system. Those four major subsystems are
- Management Controls
- Corrective and Preventive Actions (CAPA) (with satellites Medical Device Reporting, Corrections and Removals, and Medical Device Tracking)
- Design Controls
- Production and Process Controls (P&PC) (with satellite Sterilization Process Controls)
It’s important to recognize that you are responsible for ALL aspects of your quality system, its procedures and records. Even though at normal FDA inspection may focus on only these four major areas, they’re not limited to these and can ask about and review anything related to your quality system. So make sure when you conduct your internal audits, you add other subsystems as well. One approach is to perform a quarterly audit using these four subsystems as your primary topics, adding a portion (e.g., ¼) of your remaining quality system topics to each audit. In this way you will be examining and addressing issues with every part of your quality system each year.
Q. Do I have to provide the FDA inspectors everything they ask for?
A. No, not everything, as noted below in an excerpt from the QSIT, however your cooperative demeanor in working with the FDA is very important. They’re just doing their jobs. You’re required to comply with the law. To the extent you show cooperative, genuine interest in achieving both objectives, the relationship and the inspection activities will go much more smoothly.
When contacting the firm for the preannounced QSIT Inspection, the investigator should ask for a copy of the firm’s Quality Policy and high-level Quality System Procedures (including Management Review Procedures), Quality Manual, Quality Plan or equivalent documents to preview prior to the inspection. The firm is not required to supply these documents. The investigator should tell the firm that the preview of these procedural documents would facilitate the inspection. The documents would be returned at the time of the inspection. If you find deficiencies in these documents, you should request copies of the original documents after you initiate the inspection.
For each of the four major subsystems and a few satellite topics, the QSIT includes three major sections:
- Objectives
- Flow Chart
- Narrative
Below are the Objectives and Flow Charts. Prior to an inspection, or your own internal audit, you should also review the Narrative sections in the QSIT guidance document. They will provide very useful insight into the way the inspector is likely to approach the questioning.
Management Controls
Inspectional Objectives:
- Verify that a quality policy, management review and quality audit procedures, quality plan, and quality system procedures and instructions have been defined and documented.
- Verify that a quality policy and objectives have been implemented.
- Review the firm’s established organizational structure to confirm that it includes provisions for responsibilities, authorities and necessary resources.
- Confirm that a management representative has been appointed. Evaluate the purview of the management representative.
- Verify that management reviews, including a review of the suitability and effectiveness of the quality system, are being conducted.
- Verify that quality audits, including re-audits of deficient matters, of the quality system are being conducted.At the conclusion of the inspection….
7. Evaluate whether management with executive responsibility ensures that an adequate and effective quality system has been established and maintained.
Flow Chart:
See Figure 2. Note the references to the Quality System Regulation sections, 21 CFR part 820.xx. Most all of the Flow Chart blocks have such references, either to 820 or other sections of the regulation that apply (e.g., 803, 806, 821).
Figure 2.

Corrective and Preventive Actions (CAPA)
Inspectional Objectives
While CAPA is one of the four major areas of investigation, remember that most CAPA initiatives come from complaints. So, you can expect the inspector to review your complaint procedures and records as well.
- Verify that CAPA system procedure(s) that address the requirements of the quality system regulation have been defined and documented.
- Determine if appropriate sources of product and quality problems have been identified. Confirm that data from these sources are analyzed to identify existing product and quality problems that may require corrective action.
- Determine if sources of product and quality information that may show unfavorable trends have been identified. Confirm that data from these sources are analyzed to identify potential product and quality problems that may require preventive action.
- Challenge the quality data information system. Verify that the data received by the CAPA system are complete, accurate and timely.
- Verify that appropriate statistical methods are employed (where necessary) to detect recurring quality problems. Determine if results of analyses are compared across different data sources to identify and develop the extent of product and quality problems.
- Determine if failure investigation procedures are followed. Determine if the degree to which a quality problem or nonconforming product is investigated is commensurate with the significance and risk of the nonconformity. Determine if failure investigations are conducted to determine root cause (where possible). Verify that there is control for preventing distribution of nonconforming product.
- Determine if appropriate actions have been taken for significant product and quality problems identified from data sources.
- Determine if corrective and preventive actions were effective and verified or validated prior to implementation. Confirm that corrective and preventive actions do not adversely affect the finished device.
- Verify that corrective and preventive actions for product and quality problems were implemented and documented.
- Determine if information regarding nonconforming product and quality problems and corrective and preventive actions has been properly disseminated, including dissemination for management review.
Flow Chart:
See Figure 3.
 Figure 3.
Figure 3.
Conclusion
In Part 2 we’ll look at Medical Device Reporting, product Corrections and Removals, Medical Device Tracking, the very important and broadly applicable Design Controls, and Production & Process Controls including Sterilization Process Controls.
Larry Blankenship, Director, Boulder iQ