

How to survive an FDA inspection! Part 2
Surviving an FDA Inspection Part 2
In Part 1 we discussed the key logistics involved with the FDA visiting your facility and the types of inspections they might wish to perform. Initial inspections, routine inspections and Quality System Surveillance Inspections are the most common. The FDA publishes guidance documents to be used by their inspectors in conducting the inspections, so our recommendation is to get copies of those documents (all linked in Part 1) and perform your own internal inspections following the FDA’s own guidance. See part 1 for details.
Here in part 2 we complete looking into the remaining key topics for inspection including Medical Device Reporting, product Corrections and Removals, Medical Device Tracking, the very important and broadly applicable Design Controls, and Production & Process Controls including Sterilization Process Controls.
Medical Device Reporting
Inspectional Objectives
- Verify that the firm has MDR procedures that address the requirements in 21 CFR Part 803.17.
- Verify that the firm has established and maintains MDR event files that comply with 21 CFR Part 803.18.
- Confirm that the appropriate MDR information is being identified, reviewed, reported, documented and filed.
- Confirm that the firm follows their procedures and they are effective in identifying MDR reportable deaths, serious injuries and malfunctions.
Flow chart
See Figure 4.
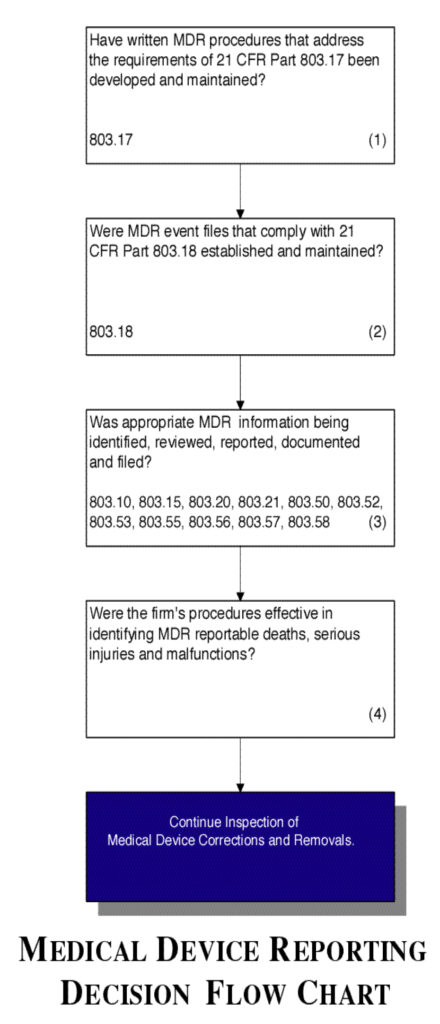
Figure 4.
Reports of Corrections and Removals
Inspectional Objectives
- Determine if corrections or removals of a device were initiated by the manufacturer.
- Confirm that the firm’s management has implemented the reporting requirements of 21 CFR Part 806.
- Verify that the firm has established and continues to maintain a file for all non-reportable corrections and removals per 21 CFR Part 806.20. Also verify that the firm is complying with the other file-related requirements of 21 CFR Part 806.
Flow chart
See Figure 5.
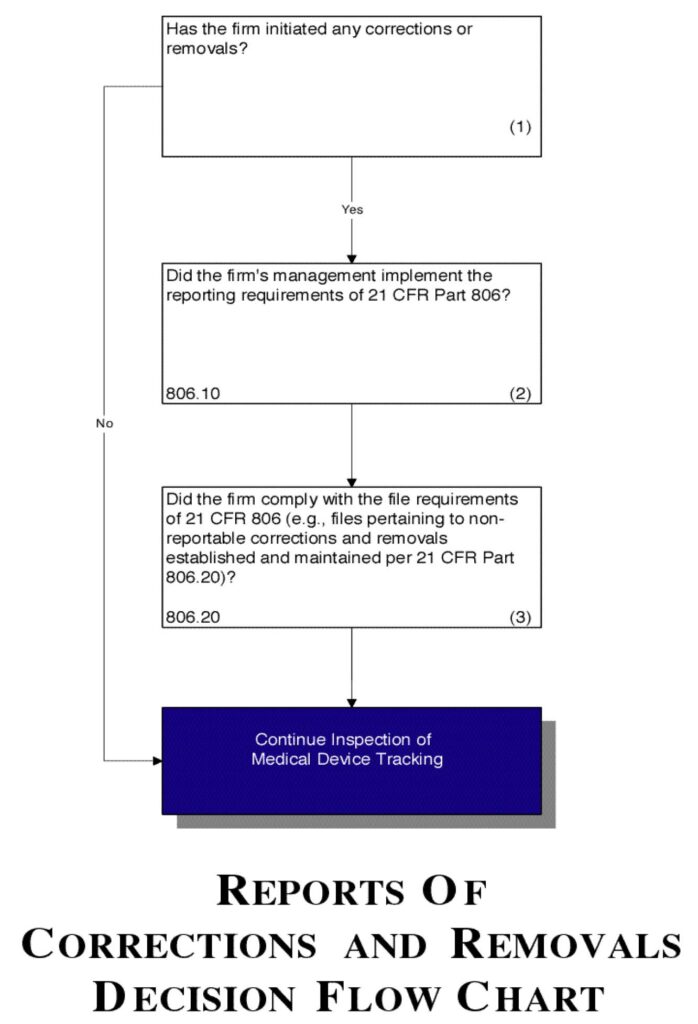
Figure 5.
Medical Device Tracking
Not all products require tracking. The FDA published a Guidance Document titled “Medical Device Tracking, Guidance for Industry and Food and Drug Administration Staff”. This document includes a list of products that must be tracked.
Inspectional Objectives
- Determine if the firm manufactures or imports a tracked device.
- Verify that the firm has established a written standard operating procedure (SOP) for tracking that complies with the requirements in 21 CFR Part 821.25(c).
- Verify that the firm’s quality assurance program includes audits of its tracking system within the appropriate timeframes specified in 21 CFR Part 821.25(c)(3).
Flow chart
See Figure 6.
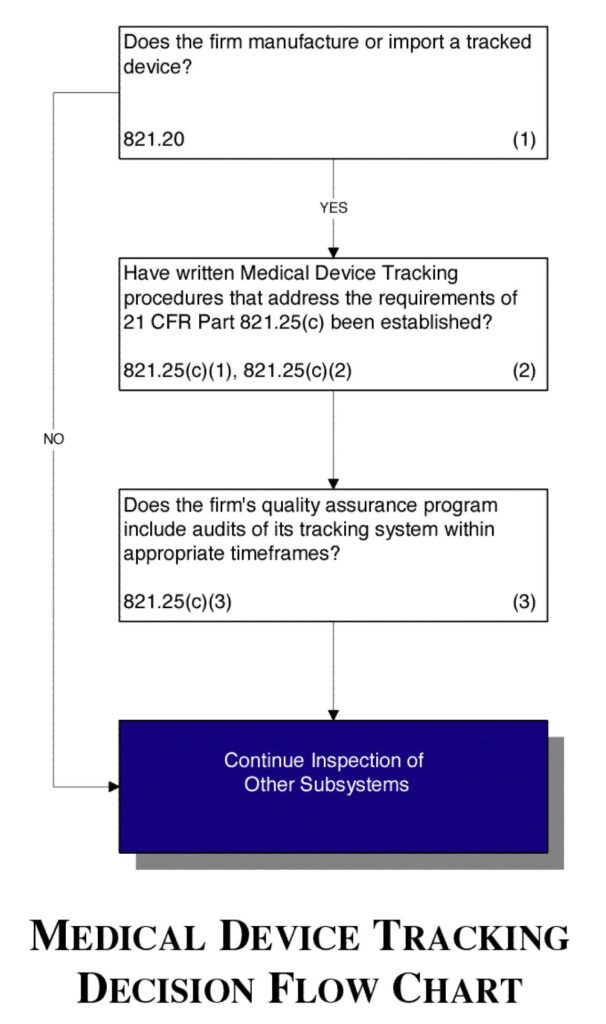
Figure 6.
Design Controls
Inspectional Objectives
- Select a single design project.
Note: If the project selected involves a device that contains software, consider reviewing the software’s validation while proceeding through the assessment of the firm’s design control system. - For the design project selected, verify that design control procedures that address the requirements of Section 820.30 of the regulation have been defined and documented.
- Review the design plan for the selected project to understand the layout of the design and development activities including assigned responsibilities and interfaces.
Note: Evaluate the firm’s conduct of risk analysis while proceeding through the assessment of the firm’s Design Control system. - Confirm that design inputs were established.
- Verify that the design outputs that are essential for the proper functioning of the device were identified.
- Confirm that acceptance criteria were established prior to the performance of verification and validation activities.
- Determine if design verification confirmed that design outputs met the design input requirements.
- Confirm that design validation data show that the approved design met the predetermined user needs and intended uses.
- Confirm that the completed design validation did not leave any unresolved discrepancies.
- If the device contains software, confirm that the software was validated.
- Confirm that risk analysis was performed.
- Determine if design validation was accomplished using initial production devices or their equivalents.
- Confirm that changes were controlled including validation or where appropriate verification.
- Determine if design reviews were conducted.
- Determine if the design was correctly transferred.
Flow chart:
See Figure 7.

Figure 7.
Production and Process Controls
Inspectional Objectives
- Select a process for review based on:
- CAPA indicators of process problems;
- Use of the process for manufacturing higher risk devices;
- Degree of risk of the process to cause device failures;
- The firm’s lack of familiarity and experience with the process;
- Use of the process in manufacturing multiple devices;
- Variety in process technologies and Profile classes; Processes not covered during previous inspections;
- Any other appropriate criterion as dictated by the assignment
Note: If the process chosen is sterilization, evaluate the process according to the “Sterilization Process Controls” chapter of this handbook.
- Review the specific procedure(s) for the manufacturing process selected and the methods for controlling and monitoring the process. Verify that the process is controlled and monitored.
Note: Control and monitoring procedures may include in-process and or finished device acceptance activities as well as environmental and contamination control measures. - If review of the Device History Records (including process control and monitoring records, etc.) reveals that the process is outside the firm’s tolerance for operating parameters and/or rejects or that product nonconformances exist:
- Determine whether any nonconformances were handled appropriately;
- Review the equipment adjustment, calibration and maintenance; and
- Evaluate the validation study in full to determine whether the process has been adequately validated.
- If the results of the process reviewed cannot be fully verified, confirm that the process was validated by reviewing the validation study.
- If the process is software controlled, confirm that the software was validated.
- Verify that personnel have been appropriately qualified to implement validated processes or appropriately trained to implement processes which yield results that can be fully verified.
Flow chart
See Figure 8.
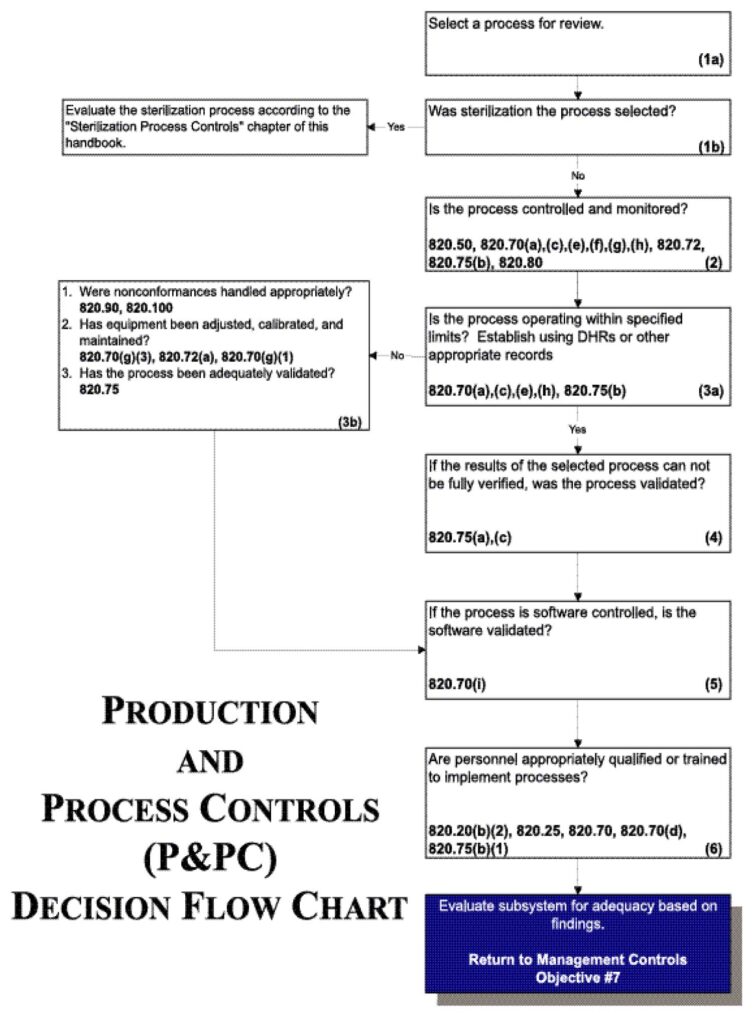
Figure 8.
Sterilization Process Controls
Inspectional Objectives
- Confirm that the sterilization process was validated by reviewing the validation study.
- Review the specific procedure(s) for the sterilization process selected and the methods for controlling and monitoring the process. Verify that the process is controlled and monitored.
- If review of the Device History Records (including process control and monitoring records, acceptance activity records, etc.) reveals that the sterilization process is outside the firm’s tolerance for operating or performance parameters:
- Determine whether the nonconformances were handled appropriately; and
- Review the equipment adjustment, calibration, and maintenance
- If the sterilization process is software controlled, confirm that the software was validated.
- Verify that personnel have been appropriately qualified and trained to implement the sterilization process.
Flow chart
See Flow Chart 9.
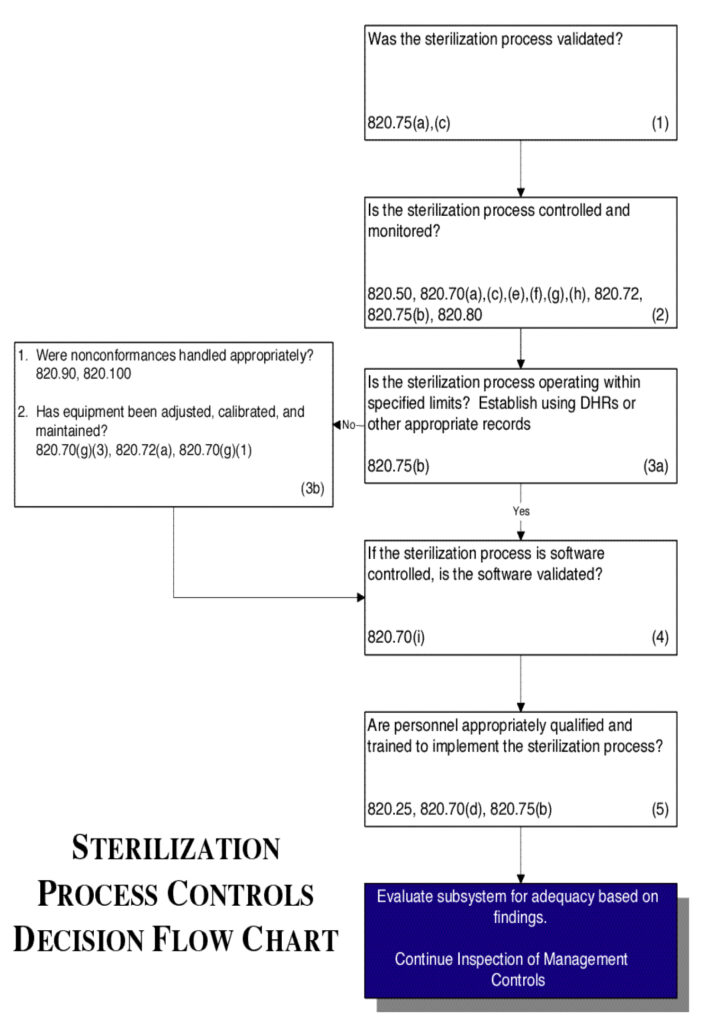
Figure 9.
So what then happens at that Friday meeting with the inspector? If the inspector has observations regarding issues that are not in compliance with the QSR, they will discuss them with you and may report them on a Form 483. The form 483 is the formal report to you and to your FDA file that the inspection revealed observations that need to be addressed. They may be minor or major in nature, and you must respond with corrections that will be verified at the next inspection if not before.
Here’s an excerpt from the IOM on this topic:
All FDA-483s should adhere to the following general principles:
- Observations which are listed should be significant and correlate to regulated products or processes being inspected.
- Observations of questionable significance should not be listed on the FDA-483 but will be discussed with the firm’s management so that they understand how uncorrected problems could become a violation.
- Each observation should be clear and specific.
- Each should be significant. Length is not necessarily synonymous with significance.
- Observations should not be repetitious.
- The observations should be ranked in order of significance.
- All copies of the FDA-483 should be legible.
Conclusion
If you’re a medical device company, especially if your product is class 2 or greater, you WILL be inspected by the FDA. Further, the inspections will probably be repeated every two years or so. Therefore, it behooves you to be prepared.
Fortunately, if you have your RA staff and QMS Quality Representative review the guidance documents used to train FDA inspectors, you can be prepared. The old adage “failing to plan is planning to fail” is very applicable here. It’s up to you. Does this mean you’ll never get a 483 observation? Of course not. One would hope implementation is perfect, but people make mistakes and different people interpret the regulations differently. But this important discipline of self-auditing and internally correcting the issues and errors you find will serve you well.
You and your company are providing important products and services to the medical community. Good for you! If you keep it up and do it right, everybody wins!
Larry Blankenship, Director, Boulder iQ